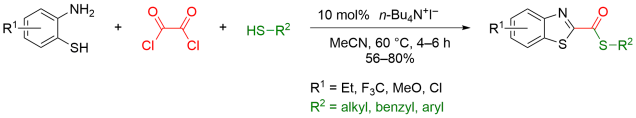
Multicomponent reaction was my research topic when I was finishing my master’s degree. While I was browsing the Journal of Organic Chemistry, I stumbled upon a multicomponent reaction reported by Abu Khan from Indian Institute of Technology Guwahati, Nahid Ali from Indian Institute of Chemical Biology, and their co-workers. They combined 2-aminothiophenols, oxalyl chloride, and thiols to form benzothiazoles with a thioester moiety at the 2-position. Tetra-n-butylammonium iodide (10 mol%) was used as a catalyst, and the optimal reaction conditions were found in acetonitrile at 60 °C. With this protocol, various benzothiazoles could be prepared in fair to good yields within four to six hours. Maximum yields were observed when both 2-aminothiophenols and thiols possessed electron-donating groups. Oxalyl chloride, tetra-n-butylammonium iodide, and different 2-aminothiophenols and thiols are commercially available.
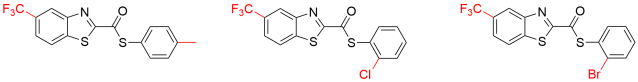
In vitro analyses by the investigators indicated that benzothiazoles below were active against Leishmania donovani, the protozoan causing visceral leishmaniasis, also known as black fever because of the darkening of the skin noticed on infected people in India.
Teleportation gates:
- Article: Dar, A. A.; Shadab, M.; Khan, S.; Ali, N.; Khan, A. T. J. Org. Chem. 2016, 81, 3149–3160. DOI: 10.1021/acs.joc.6b00113.
- Indian Institute of Technology Guwahati.
- Abu Khan’s research group.
- Indian Institute of Chemical Biology.
- Nahid Ali’s research group.
- Leishmania donovani on Wikipedia.
- Visceral leishmaniasis on Wikipedia.












{kind=link}